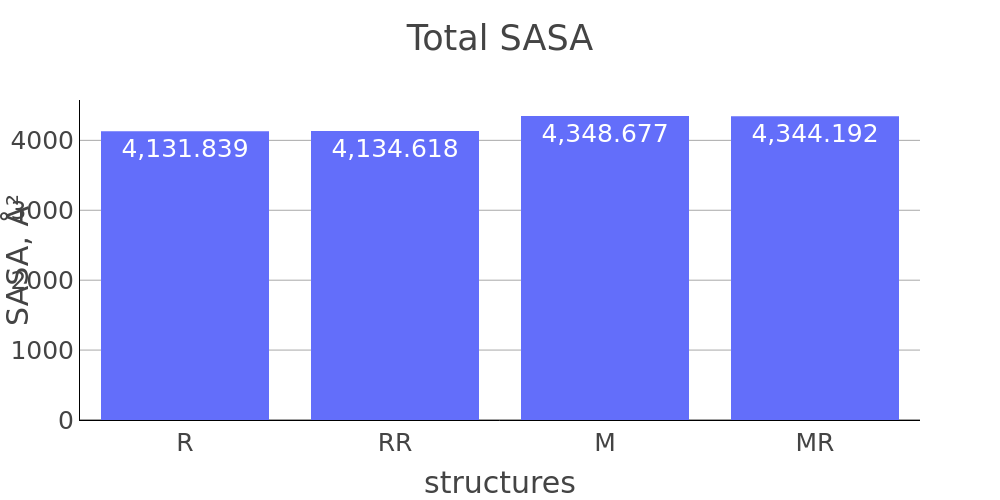
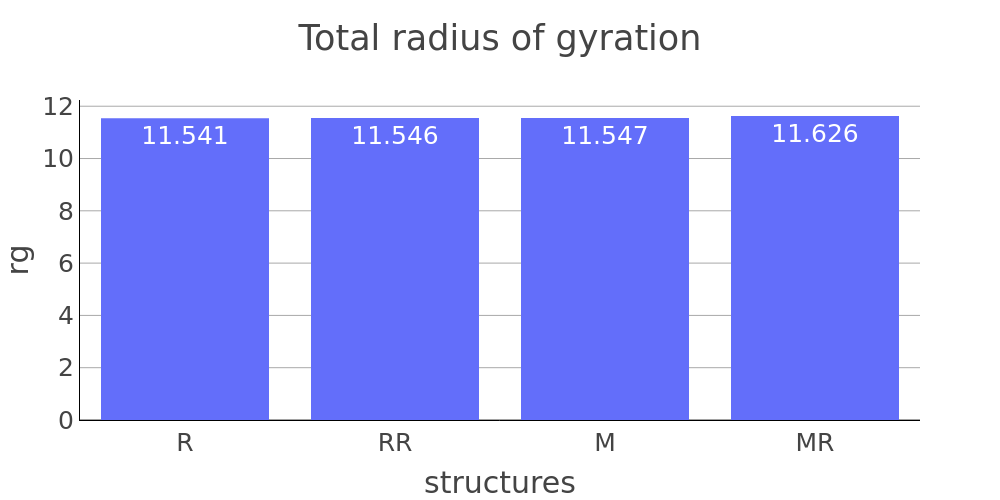
Report description
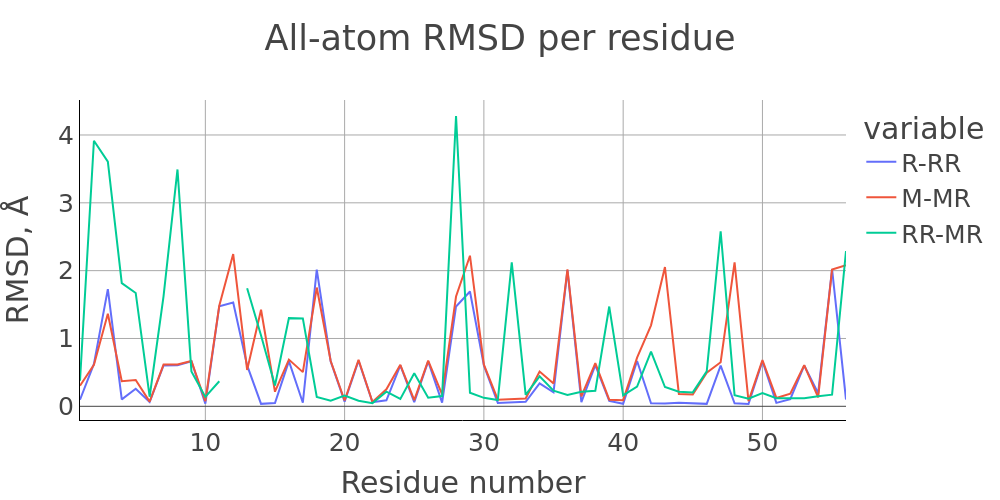
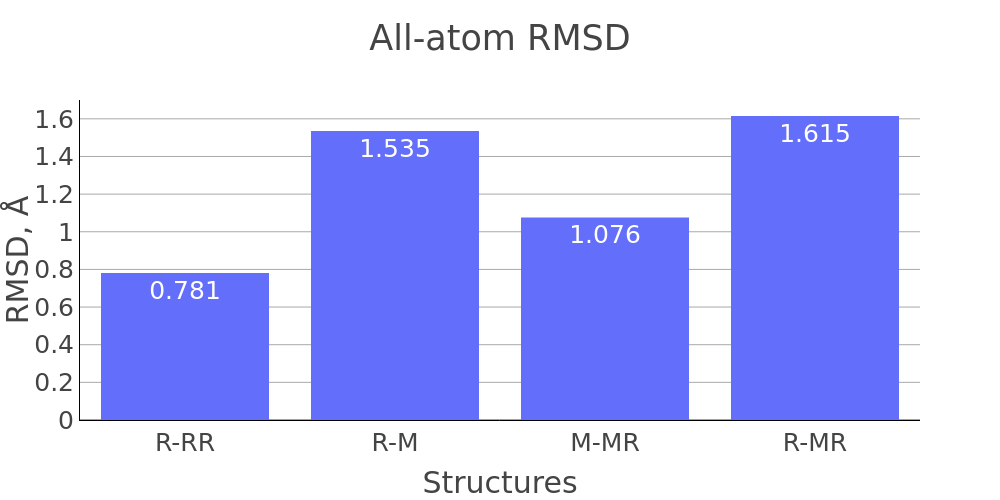
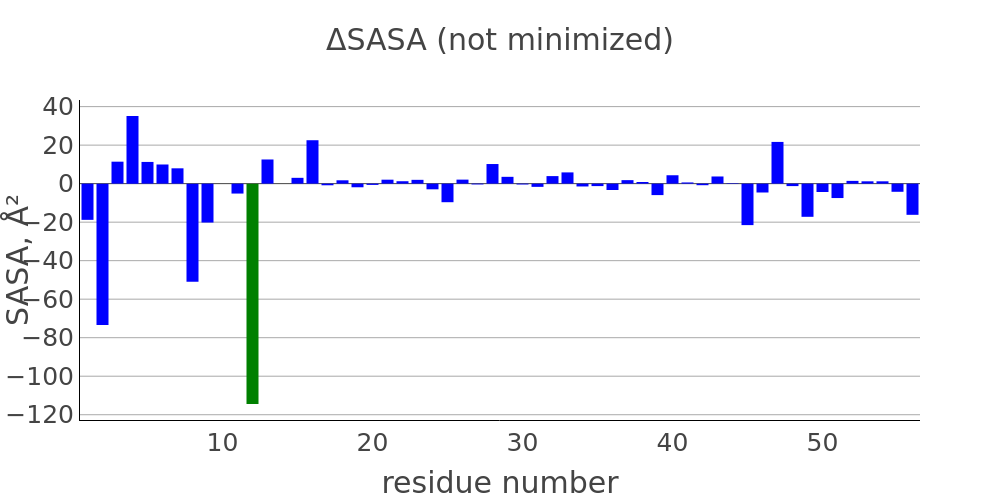
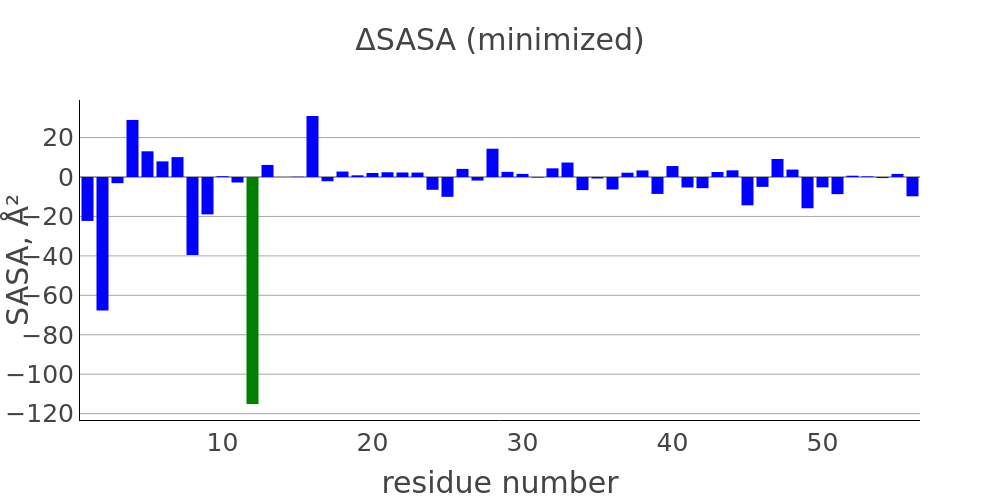
The report presented above includes plots showing changes of protein chain geometrical indicators as a result of modification. Results are whown for four versions of structure marked as in list below:
- R: reference. Intact structure without modification
- RR: reference relaxed. Intact structure after energy minimization (EM)
- M: Modified. Structure with PTM before EM
- MR: modified relaxed. Structure with PTM after EM
Algorithms used to predict the results are:
SCPacker: predicting side-chains position after modification
Petrovskiy, D.V.; Nikolsky, K.S.; Rudnev, V.R.; Kulikova, L.I.; Butkova, T.V.; Malsagova, K.A.; Kopylov, A.T.; Kaysheva, A.L. Modeling Side Chains in the Three-Dimensional Structure of Proteins for Post-Translational Modifications. Int. J. Mol. Sci. 2023, 24, 13431. https://doi.org/10.3390/ijms241713431
GROMACS energy minimisation steepest decent algorithm. Details: https://manual.gromacs.org/.../energy-minimization.html. Cite: https://manual.gromacs.org/.../preface.html
Calculated values uses hydrogen placing algorythm implemented in PyMol: https://pymol.org/